Terminology
We will be using examples associated with genomic analyses in bioinformatics, therefore, it is important for you to become familiar with the following terms.
What is a read?
A read is a string of bases represented by their one letter codes. Here is an example of a read that is 100 bases long. TTAACCTTGGTTTTGAACTTGAACACTTAGGGGATTGAAGATTCAACAACCCTAAAGCTTGGGGTAAAACCTTAGGGGAT
What is a contig?
A contig is the consensus sequence generated by aligning reads to themselves.
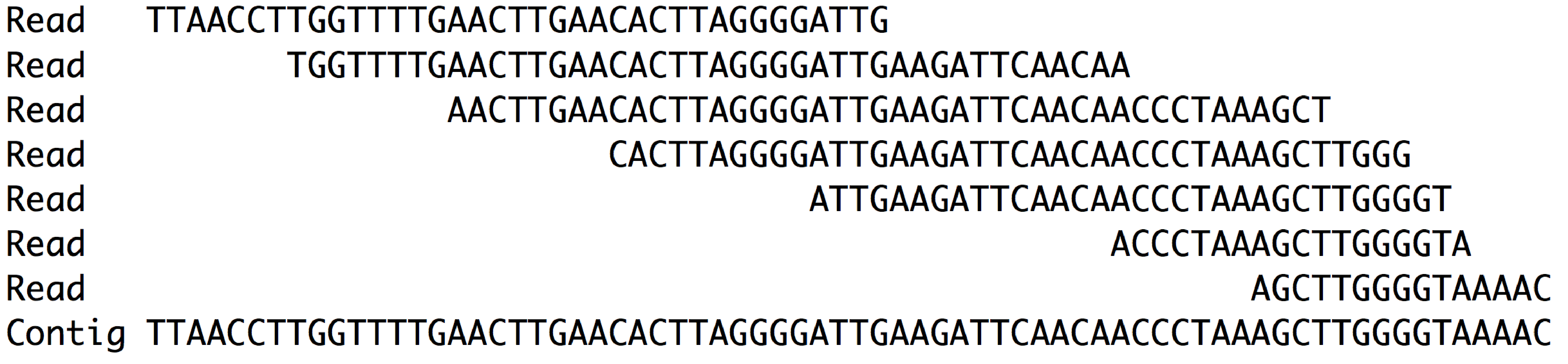
Alignment and Consensus
The last line is the consensus of the aligned reads which we call a contig.
What is a scaffold?
A scaffold is a set of contigs that have been ordered and oriented based on mate pair or long distance information. Usually there are large gaps found between contigs. New sequencing technologies are helping to close those gaps.
What is a chromosome?
Chromosomes are the largest DNA molecules in the cell. Scaffolds can be ordered and oriented using a genetic map or HiC data into linkage groups or chromosomes. The ultimate goal of a genome assembly project is to assemble reads into chromosomes.
Next Generation Sequencing
Next-generation sequencing generates masses of DNA sequence data that’s richer and more complete than is imaginable with Sanger sequencing. Illumina sequencing systems can deliver data output ranging from 300 kilobases up to multiple terabases in a single run, depending on instrument type and configuration.
Sequence Assembly
Sequence assembly refers to aligning and merging fragments from a longer DNA sequence in order to reconstruct the original sequence. The assembly can be found at different stages of completion. It could be conting, scaffold or even chromosome level.
Sequence Annotation
DNA annotation or genome annotation is the process of identifying the locations of genes and all of the coding regions in a genome and determining what those genes do.

Genes and Genome
What is a genome?
A genome is an organism’s complete set of DNA, including all of its genes. Most of the genomes available online have been partially assembled. We will find out how to retrieve these genomes and analyze them.
We will practice with other researcher’s data to become a pro. Then we can analyze our own data.